
01
Description
More about
02
Short list of features
More about
03
Technical Description
More about
04
Repositories and Download
More about
Starting in 2005, the BigDFT EU project aimed to test the advantages of Daubechies wavelets as a basis set for DFT using pseudopotentials. This led to the creation of the BigDFT code, which has optimal features of flexibility, performance and precision. In addition to the traditional cubic-scaling DFT approach, the wavelet-based approach has enabled the implementation of an algorithm for DFT calculations of large systems containing many thousands of atoms, with a computational effort which scales linearly with the number of atoms. This feature enables electronic structure calculations of systems which were impractical to simulate even very recently.
BigDFT has rapidly become a mature and reliable package suite with a wide variety of features, ranging from ground-state quantities up to potential energy surface exploration techniques. BigDFT uses dual space Gaussian type norm-conserving pseudpotentials including those with non-linear core corrections, which have proven to deliver all-electron precision on various ground state quantities. Its flexible poisson solver can handle a number of different boundary conditions including free, wire, surface, and periodic. It is also possible to simulate implicit solvents as well as external electric fields. Such technology is employed for the computations of hybrid functionals and time-dependent (TD) DFT.
Diffusion
BigDFT is free and open source software, made available under the GPL license. The code is developed by few individuals, ranging from 6 up to 15 people. The active code developers are, or have been, located in various groups in the world, including EU, UK, US, and Japan. For these reasons, in addition to production calculations aimed at scientific results, the code has been often employed as a test-bed for numerous case-study in computer science and by hardware/software vendors, to test the behaviour of novel/prototype computer architectures in realistic runtimes.
The compilation of the code suite relies on the splitting of the code components into modules, which are compiled by the \texttt{bundler} package. This package lays the groundwork for developing a common infrastructure for compiling and linking together libraries for electronic structure codes, and it is employed as the basis for the ESL bundle.
Performance in HPC environments
BigDFT is a award-winner DFT code, recipient of the first edition (2009) of the Bull-Fourier prize for its "the ground-breaking usage of wavelets and the adaptation to hybrid architectures, combining traditional processors and graphic accelerators, leading the path for new major advancements in the domain of new materials and molecules". It is parallelized using a combination of MPI and OpenMP and has support for GPU acceleration since the early days of GPGPU computing. Such supports involve both CUDA as well as OpenCL computing kernels, and can be routinely applied to large systems. For example, the calculation of a 12,000 atom protein system requires about 1.2 hours of wall-time on 16 nodes of the Irene-ROME supercomputer. This calculation can be further accelerated for systems composed of repeated sub-units using a fragment approach for molecules, or in the case of extended systems, a pseudo-fragment approach, both among the outcomes of MaX2 project.

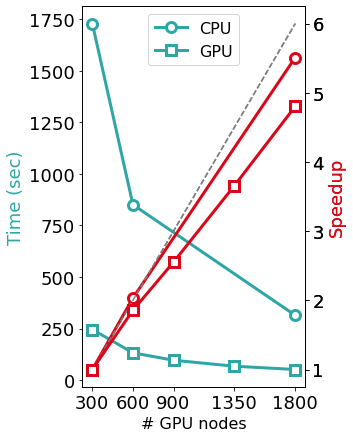
The Figure above shows the benefits induced by a multi-GPU calculation on a PBE0 calculation (seconds per 2 SCF iterations) of a system made of 5400 KS orbitals on Piz Daint. Such calculations can scale effectively up to the range of thousands of GPUs and compute nodes. To facilitate driving the calculations of dense workflow graphs involving thousands of simulations of large systems, the code suite includes a python package called PyBigDFT as a framework for managing DFT workflows. PyBigDFT is able to handle building complex systems or reading them from a variety of file types, performing calculations with BigDFT linked with AiiDA package, and analyzing calculation results. This makes it easy to build production analysis, thereby enabling new users' production HPC experiences on top of the data generated from large scale DFT calculations.
Currently, BigDFT is being deployed on large HPC machines including Fugaku (RIKEN, JP), Archer2 (Edinburgh, UK), and Irene-ROME (TGCC-CEA, FR).
Under construction.
Software
BigDFT incorporates a hybrid MPI/OpenMP parallelization scheme, with parts of the code also ported to GPUs. For both the original cubic scaling version of BigDFT and the more recently developed localized support function approach, the MPI parallelization is at the highest level with orbitals, or in the latter case support functions, divided between MPI tasks. Two different data layouts are used to divide the orbitals/support functions among the tasks depending on the operation involved, such that each task is either responsible for a fixed number of orbitals, or each task is responsible for all orbitals which are defined over a given set of grid points. OpenMP is then used to parallelize operations at a lower level, for example in the calculation of the charge density and when generating the overlap and Hamiltonian matrices.
I/O requirements
The largest data files output by BigDFT are those containing the support functions. Thanks to the strict localization of these support functions, the file size remains constant as the system size increases (approximately 1 MB per file), and thanks to the fragment approach, the number of files should also not increase significantly so that even very large systems will have small data requirements. We therefore also have minimal I/O requirements: for BigDFT, I/O is limited to the start and end of a calculation, where there is reading and writing of the support functions to disk. As mentioned above, the size and number of files will not (or only to a very limited extent) increase with system size. Currently there maybe multiple MPI tasks reading from disk; so far this has not proven to be a bottleneck, but should this prove to be a problem when running at scale it would be straightforward to modify the code so that only one MPI task reading each file and communicating the data. Due to the small file sizes of the generated data, analysis will not require significant computational resources and will be limited to tasks such as calculating pair distribution functions and analyzing atomic coordination. These can either be performed as a post-processing step on a desktop computer or during the simulation. Likewise, we have not got an intensive workflow scheme, as the fragment approach merely requires the small 'template' calculation to be run once following which multiple large calculations can be run. However, as discussed above, we will make use of scripting to couple together runs e.g. for different temperatures and different defect types
Parallel programming
BigDFT use a hybrid MPI/OpenMP parallelization scheme. The use of a hybrid approach allows good scaling behaviour to be achieved on different architectures for both versions of BigDFT, and in the case where memory is a limiting factor, the usage of OpenMP ensures that all processors on a given node are performing computational work. Further details on the parallelization scheme are given elsewhere.