
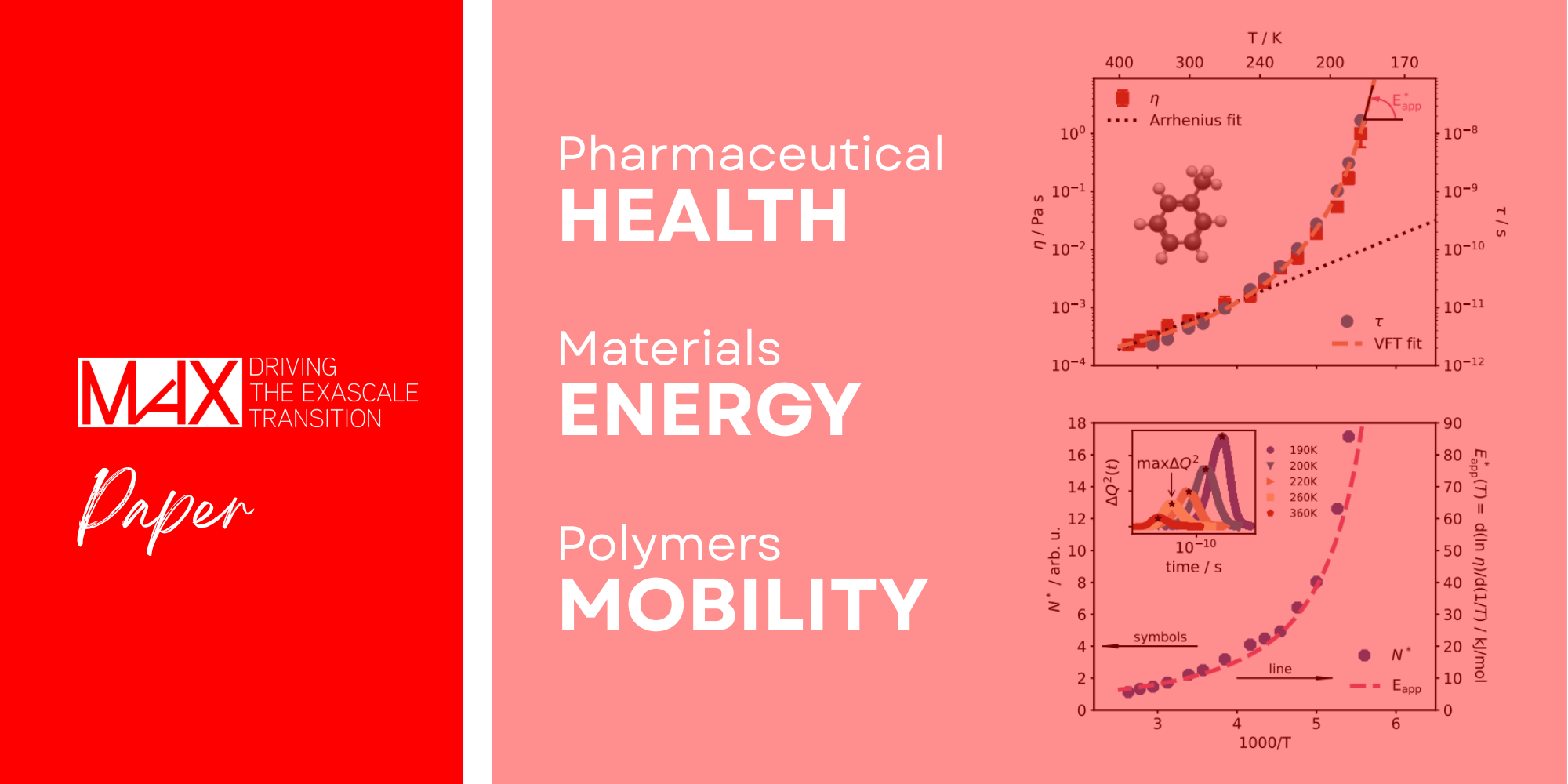
MaX researchers performed microsecond, first-principles simulations of glass-forming toluene by integrating Quantum ESPRESSO with machine-learned interatomic potentials. The researchers demonstrated that the temperature-dependent increase in apparent activation energy is proportional to the growth of dynamically correlated molecular regions near the glass transition.
Application sectors: Health (pharmaceutical stability), Energy (advanced materials), Mobility (polymers and lightweight components).
Keywords: Density Functional Theory, Neural Network Potentials, Glass Transition, Toluene, High-Performance Computing.
Glasses are all around us: in packaging, coatings, polymers, and electronic materials. Unlike crystals, they lack long-range molecular order. As a liquid cools, its viscosity gradually increases until it behaves like a solid, a process known as the glass transition. Yet, despite its ubiquity, the microscopic origin of the increase in viscosity has remained elusive.
At high temperatures, molecular motion follows a simple Arrhenius behaviour, with a roughly constant activation energy. But as the liquid cools, this picture breaks down: viscosity rises much faster than expected, and the apparent activation energy grows. One long-standing hypothesis, dating back to Adam Gibbs, suggests that molecules move in increasingly large, correlated groups as temperature decreases. The size of these groups is quantified by the number of dynamically correlated molecules, N*. Directly observing and measuring N* in realistic molecular systems, however, has been a major challenge.
Most computational studies to date have relied on simplified Lennard-Jones models. While these models provide valuable insight, they fail to capture the complexity of real molecular liquids. Ab initio molecular dynamics (AIMD) based on Density Functional Theory (DFT) offers first-principles accuracy, but is limited to extremely short timescales, far too short to reach the deeply supercooled regime where glassy dynamics emerge.
To overcome this challenge, the research team combined DFT with machine-learning-accelerated simulations of toluene. This approach allowed them to extend simulations to previously unreachable timescales and observe glassy behaviour in a realistic liquid. Their results revealed a smooth increase in the apparent activation energy from the boiling point down to low temperatures, without any sharp transition between “normal” and “glassy” regimes.
Crucially, the study showed that the apparent activation energy is directly proportional to N*. In the simulations, N* grows from roughly 1 near the boiling point to about 17 at the lowest temperatures studied. Extrapolating to the experimental glass transition suggests around 43 dynamically correlated molecules. This provides evidence that the dramatic rise in viscosity is intimately linked to the cooperative motion of molecular groups, offering new microscopic insight into the glass transition. This research improves our understanding of the glass transition by directly connecting viscosity growth to collective molecular motion.
Method and computing resources used
The study combines DFT with machine learning. Reference data were generated using MaX lighthouse code Quantum ESPRESSO. The RPBE functional with D3(BJ) dispersion corrections ensured accurate intermolecular interactions. A neural network potential was trained using the Deep Potential Generator workflow. The final model was trained on 9,558 DFT configurations.
The trained model enabled molecular dynamics simulations up to 0.5 microseconds for systems of up to 512 molecules (7,680 atoms). This is roughly three orders of magnitude longer than conventional ab initio simulations. High-performance computing (HPC) resources were essential for generating the DFT dataset, training the neural networks, and running long production simulations.
In particular, MaX sofwtare Quantum ESPRESSO provided:
- Accurate electronic structure data for training
- Efficient parallel performance on HPC systems
- Reliable treatment of dispersion interactions
MaX expertise in electronic-structure simulations ensured high-quality reference data. Careful convergence, validated pseudopotentials, and scalable workflows were critical. By coupling MaX DFT simulations with machine learning, the team extended first-principles accuracy to experimentally relevant time and temperature ranges. This approach transforms electronic-structure calculations into predictive tools for complex dynamics.
Enabling realistic, predictive glass simulations
To achieve these results, the team carried out thousands of DFT calculations to train a high-accuracy neural network potential. Simulating long trajectories for large molecular systems was essential to capture reliable dynamical observables and probe the glassy behavior of toluene over microsecond timescales. The technological breakthrough of this work lies in combining first-principles electronic structure calculations with scalable machine learning, allowing simulations of complex dynamics that were previously out of reach. This approach effectively bridges the gap between atomistic theory and experimental observation.
Understanding glassy dynamics is essential for:
- Designing stable pharmaceutical formulations (Health).
- Developing durable polymers and coatings (Mobility).
- Engineering advanced amorphous materials (Energy).
From a scientific perspective, the study provides a quantitative link between the growth of viscosity and the collective motion of molecules in a realistic liquid. By demonstrating that such slow, cooperative dynamics can now be simulated with first-principles accuracy, MaX establishes a new benchmark for predictive modeling of complex materials.
Talk to us to explore how MaX codes can support your materials simulations. See the code and learn how Quantum ESPRESSO and machine learning workflows can accelerate your research.
Reference paper
Glassy dynamics in a glass-forming liquid: A first-principles study of toluene.