

01
Description
More about
02
Short list of features
More about
03
Technical Description
More about
04
Repositories and Download
More about
Quantum ESPRESSO (Quantum opEn-Source Package for Research in Electronic Structure, Simulation, and Optimisation) is a suite of applications for ab-initio electronic structure calculations using plane waves and pseudopotentials. The code is mostly written in modern Fortran.
The suite supports standard GGA and many advanced functionals such as non-local vdW-enabled, meta-GGA, Hubbard-corrected, hybrid functionals. It can be linked with Libxc and use all functionals implemented there. The codes of the suite may perform many types of ground-state calculations: self-consistent energies, forces and stresses, structural optimization, molecular dynamics (PW and CP); search for transition pathways (NEB). The suite also contains a rich apparatus of routines and methods for the computation of the linear response to external perturbations. These are used for computing dielectric responses and vibrational spectra (PHonon); optical, magnons, and EELS spectra, using time-dependent DFT (TDDFPT); electron-phonon coupling coefficients and related properties (EPW); self consistent Hubbard correction parameters (HP), and more. Quantum ESPRESSO applications are also used as starting point for workflows that implement advanced methodologies such as MBPT, QMC, DMFT and others.
- Diffusion
- Performance in HPC environments
Diffusion
Quantum ESPRESSO is released under the GPL licence. It has a sizeable basis of users, as apparent from the number of citations of the two documenting papers of 2009 [1] (16,000+) and 2017 [2] (2,500+), authored by 25,000+ scientists and by the number of messages exchanged on the users' mailing list (2000+/year). In addition to the inner circle of developers (~20), Quantum ESPRESSO often receives contributions from external users and developers. There are currently 107 forks of the main GitLab repository, and 243 of the GitHub mirror. We are aware of at least 12 independent successful software projects that rely entirely on Quantum ESPRESSO technology and codebase for further development.
Performance in HPC environments
Most of the applications of the suite are designed for the efficient usage of the state-of-the-art HPC machines using multiple parallelization levels. The basal workload distribution can be done using MPI + OpenMP multithreading, or offloading it to GPGPUs, depending on the nodes' architecture. This parallelization level also provides an efficient data distribution among the MPI ranks. This allows to compute systems with up to ~10^4 atoms. The offloading or the usage of a growing number of MPI ranks are able to scale down the computational cost of 3D FFTs and other operations on 3D data grids.
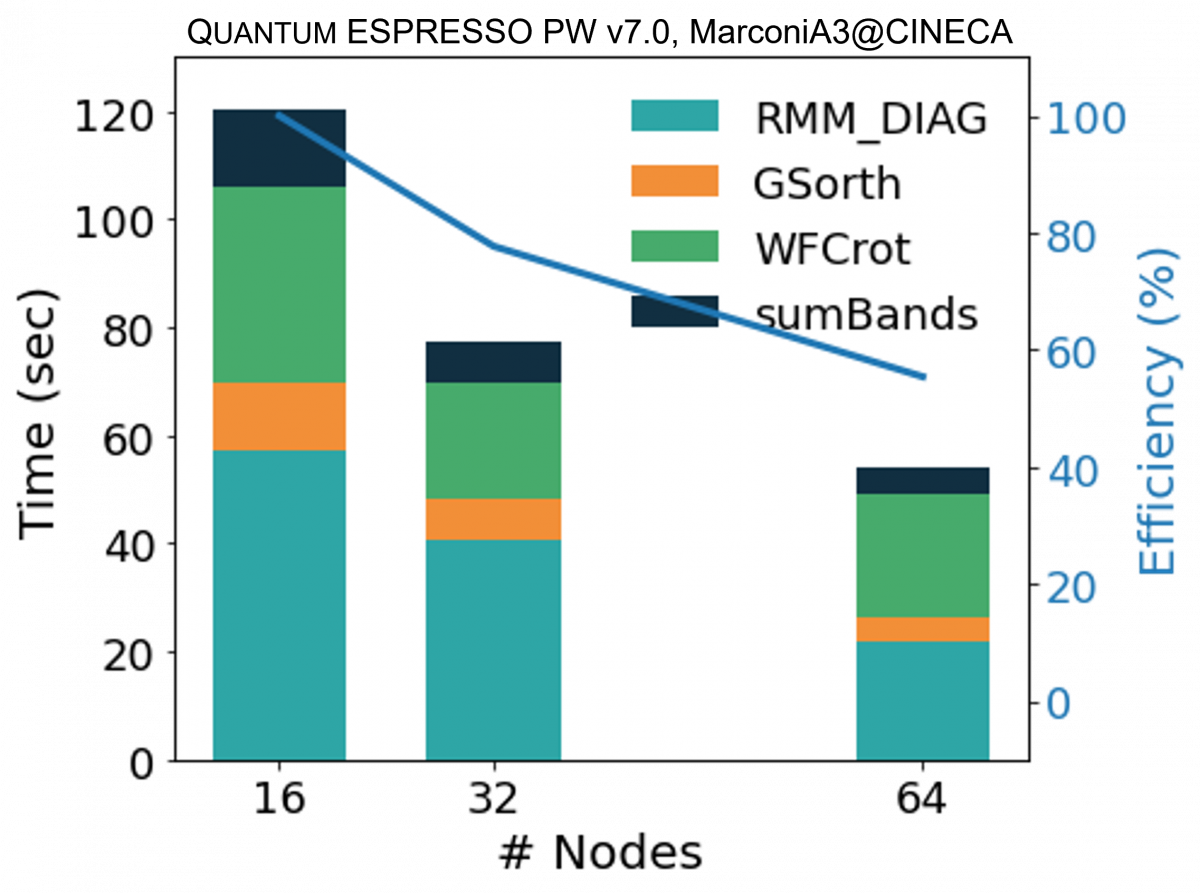
In the figure above we show the performance analysis for a mid-size case (a Carbon nanotube functionalized with two porphyrine molecules, about 1500 atoms, 8000 bands, 1 k-point) on an HPC homogeneous nodes' cluster. The kernels distribute their workload on the lower parallelization group, except for WFCrot whose perfomance relies instead on parallel or accelerated linear algebra specific libraries.
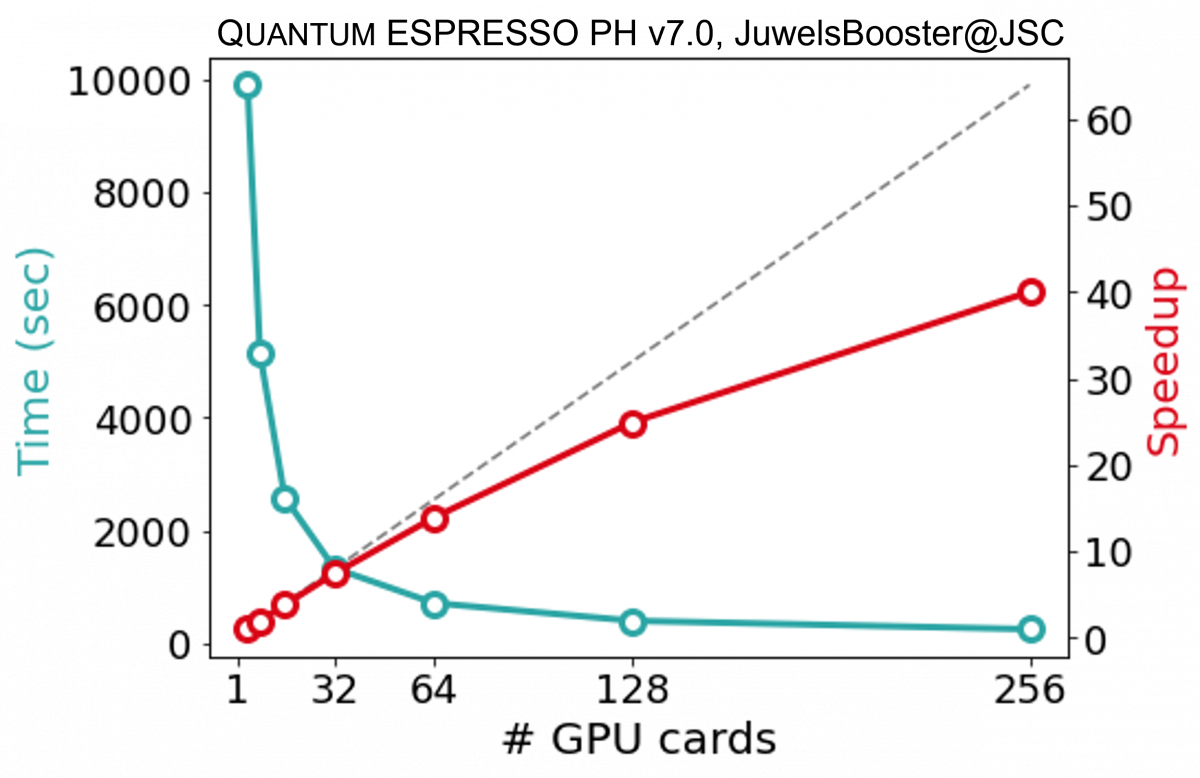
The auxiliary MPI parallelization levels allow to obtain further scaling. The band parallelization level distributes the operations on wave functions of different basal groups. The two upper parallelization levels -- pools and images -- are very efficient because they distribute the computations in concurrent quasi-independent blocks; as shown in the figure below for a PH calculation on 72 atoms quartz, executed on an heterogeneous nodes' HPC cluster equipped with Ampere GPGPUs.
References
- [1] P. Giannozzi, S. Baroni, N. Bonini et al: Quantum ESPRESSO: a modular and open-source software project for quantum simulations of materials. Journal of Physics: Condensed Matter, 21, 39, 2009.
- [2] P. Giannozzi, O. Andreussi, T. Brumme, O. Bunau, M. B. Nardelli, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, et al., Advanced capabilities for materials modelling with Quantum ESPRESSO, J. Phys.: Condens. Matter 29, 465901 (2017).
Quantum ESPRESSO is a suite of open-source codes for quantum materials modelling using the plane-wave pseudopotential method. It performs ab-initio calculations based on density-functional theory (DFT) and more advanced levels of theory: DFT+U, hybrid functionals, various functionals for van der Waals forces, many-body perturbation theory, adiabatic-connection fluctuation-dissipation theory. Within the many features available using Quantum ESPRESSO programs we mention:
- PWscf: structural optimisation and molecular dynamics on the electronic ground state, with self-consistent solution of DFT equations
- Total energy, forces, stress
- Collinear and noncollinear magnetism, spin-orbit coupling
- Structural optimisation and Born-Oppenheimer molecular dynamics;
- Berry-Phase
- CP: Car-Parrinello molecular dynamics;
- PWneb: reaction pathways and transition states with the Nudged Elastic Band method;
- PHonon: vibrational and dielectric properties from Density-Functional Perturbation Theory;
- EPW: calculation of electron-phonon coefficients in metals;
- GWL: many-body perturbation theory in the GW approach using ultra-localised Wannier functions and Lanczos chains.
Software
Quantum ESPRESSO shows a very good scalability by exploiting a hybrid MPI-OpenMP paradigm. Quantum ESPRESSO (more specifically PWscf) is parallelized on different levels: k-points (linear scaling with the number of processors), bands, and plane waves/real space grids (reaching high cpu scaling and memory distribution). Custom (domain specific) FFT’s are implemented and parallelized over planes or sticks and implement task group techniques. Parallel dense linear algebra is also exploited to improve scalability and memory distribution. Thanks to this multi-level parallelism both computation and data structures are distributed in order to fully exploit massively multi-core parallel architectures.
Particular Libraries
Quantum ESPRESSO is an open source codes distributed under GNU General Public Licence. The code source is hosted on the Github hosting service. It works with Fortran and C compilers and take advantage of highly optimized library such as BLAS, LAPACK, SCALAPACK, FFTW3, HDF5, GDLib and GSL.
Parallel programming
Two different parallelization paradigms are currently implemented in Quantum ESPRESSO, namely MPI and OpenMP. MPI is a well-established, general-purpose parallelization scheme. In Quantum ESPRESSO several parallelization levels, specified at run-time via command-line options to the executable, are implemented with MPI. This is the first choice for execution on a parallel machine. OpenMP can be implemented via compiler directives (explicit OpenMP) or via multithreading libraries (library OpenMP). Explicit OpenMP requires compilation for OpenMP execution; library OpenMP only requires linking to a multithreading version of mathematical libraries, e.g.: ESSLSMP, ACML MP, MKL (the latter is natively multi-threading).
I/O requirements
Electronic structure data, like for instance the wavefunctions, are written and read by Quantum ESPRESSO PWscf executable using a direct access to file: each task uses a raw format to store its information. These files can be used to produce checkpoints (restart).