
Machine Learning: Improved predictions for time-to-solution of materials science simulations

The rise of hybrid systems, where the standard central processing unit (CPU) is accompanied by one or more accelerators, and the drastic increase in the number of cores per node requires the adoption of different algorithmic strategies for the same problem and inflates the number of options for parallel or accelerated execution of scientific codes. This, in turn, makes the (parallel) execution of such applications more complex and their performance harder to predict.
The primary effect today from the user standpoint is that sub-optimal execution schemes are often adopted since a complete exploration of the complicated and interdependent set of execution options is a lengthy and hardly automatable task.
In order to tackle this problem, in a recently published paper by Pittino et al, machine learning techniques are used to predict the time required by the large set of algorithms utilized in a self-consistent field iteration of the Density Functional Theory method, using only input details file and runtime options as descriptors.
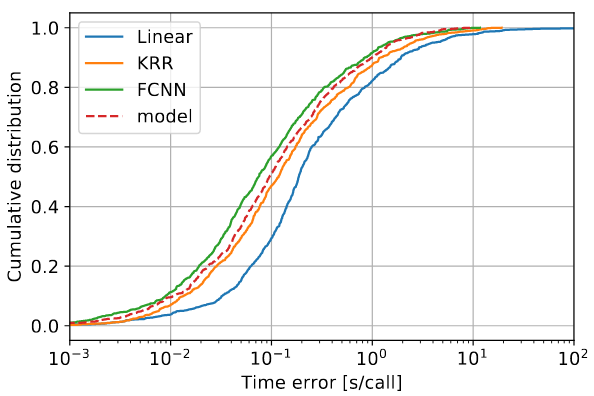
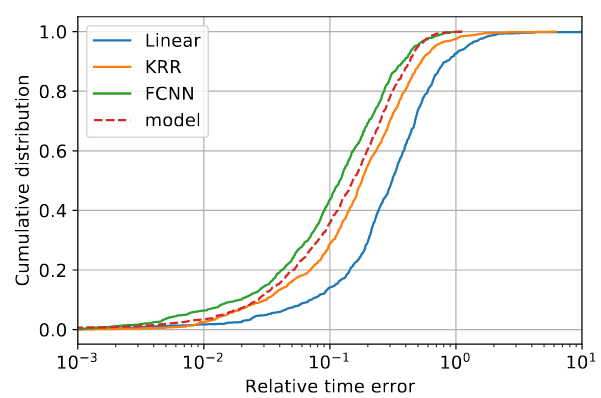
Distribution of the timing errors (absolute and relative) for three different algorithms - Linear Regression, Kernel Ridge Regression (KRR), Fully Connected Neural Network (FCNN) - compared to the analytical model.
As expected, the best performance is obtained by using advanced neural network techniques, which are found to even beat the accuracy of a full-custom analytical model specifically derived using expert knowledge of the application, improving the prediction error by up to 5%.
These results represent an important step towards the construction of accurate tools for optimal scheduling of computational simulations and identification of execution issues.