
11 July 2024
Theoretical Analysis of Electronic and Optical Properties in Bulk and Monolayer BiI3 for Optoelectronic Applications

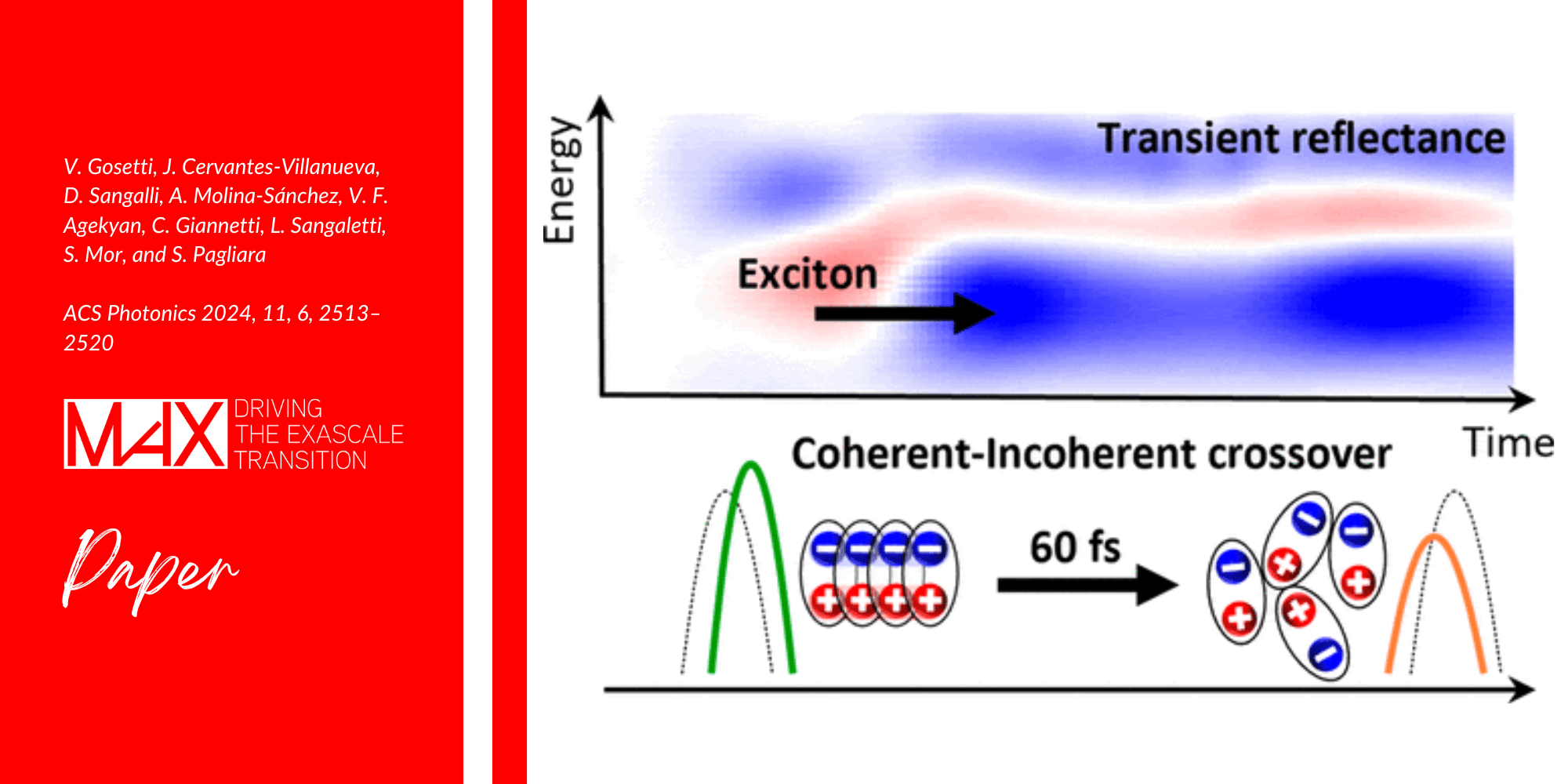
A team of researchers propose a detailed theoretical study of the electronic and optical properties of bulk and monolayer bismuth triiodide (BiI3) including the effects of spin-orbit coupling and semi-core states.
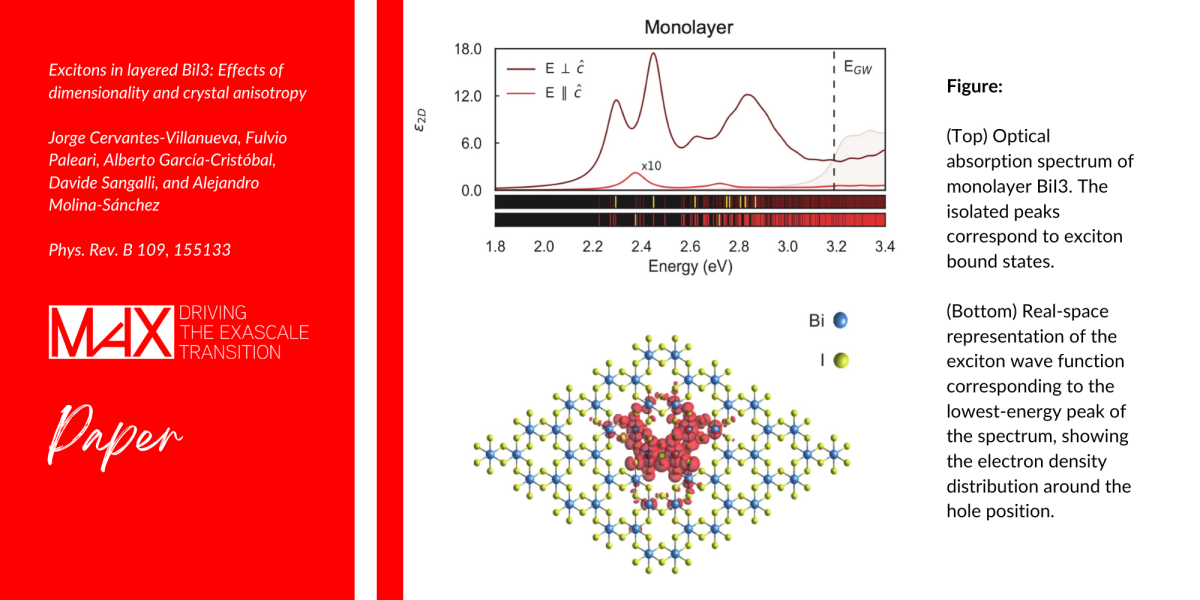
Bismuth triiodine (BiI3) is an important member of the trihalide family of layered materials. Experimental studies indicate that it is a prime candidate to explore the ultrafast dynamics of electronic excitations, which is dominated by electron-hole bound pairs (excitons). However, the excited-state properties of this material were never studied in detail.
This work provides the first accurate study of excitons in bulk and monolayer BiI3, fully including the effects of spin-orbit coupling and semi-core states. In particular, the researchers illustrate the physics of the energy splitting between longitudinal and transverse excitations.
To carry out their study, the researchers benefited from two of the MaX lighthouse codes:
<<We used the Quantum ESPRESSO code to compute the ground-state properties of the system within density functional theory, and the YAMBO code to compute quasiparticle corrections and to solve the Bethe-Salpeter equation for excitons within many-body perturbation theory. In particular, we calculated exciton dispersion relations including the nonanalytic contribution induced by the long-range Coulomb interaction.>> says Fulvio Paleari (CNR-Nano, Italy), one of the authors of the scientific paper.
The results achieved by the team of researchers provide a reference for future experimental measurements. The interplay between spin-orbit coupling and large binding energy, together with the role of quantum confinement, confirm that BiI3 is an interesting material for optoelectronic applications and show that it is a good candidate for the study of exciton dynamics.
About Quantum ESPRESSO
Quantum ESPRESSO is the major open-source (set of) code(s) for quantum materials modelling using the plane-wave pseudopotential method; it has been the development platform for such important methodological innovations as Car-Parrinello molecular dynamics and Density-Functional Perturbation Theory.
About YAMBO
YAMBO is an open-source code that implements Many-Body Perturbation Theory (MBPT) methods (such as GW and BSE), which allows for accurate prediction of fundamental properties as band gaps of semiconductors, band alignments, defect quasi-particle energies, optics and out-of-equilibrium properties of materials, including nano-structured systems.
Reference article:
Excitons in layered BiI3: Effects of dimensionality and crystal anisotropy.