
27 May 2024
Origins of spin excitation gap in CrI3 quasi-two-dimensional single crystals unveiled through advanced calculations

By performing first-principles calculations based on time-dependent density functional perturbation theory, a team of researchers show the nature of the observed gap in CrI3 quasi-two-dimensional single crystals.
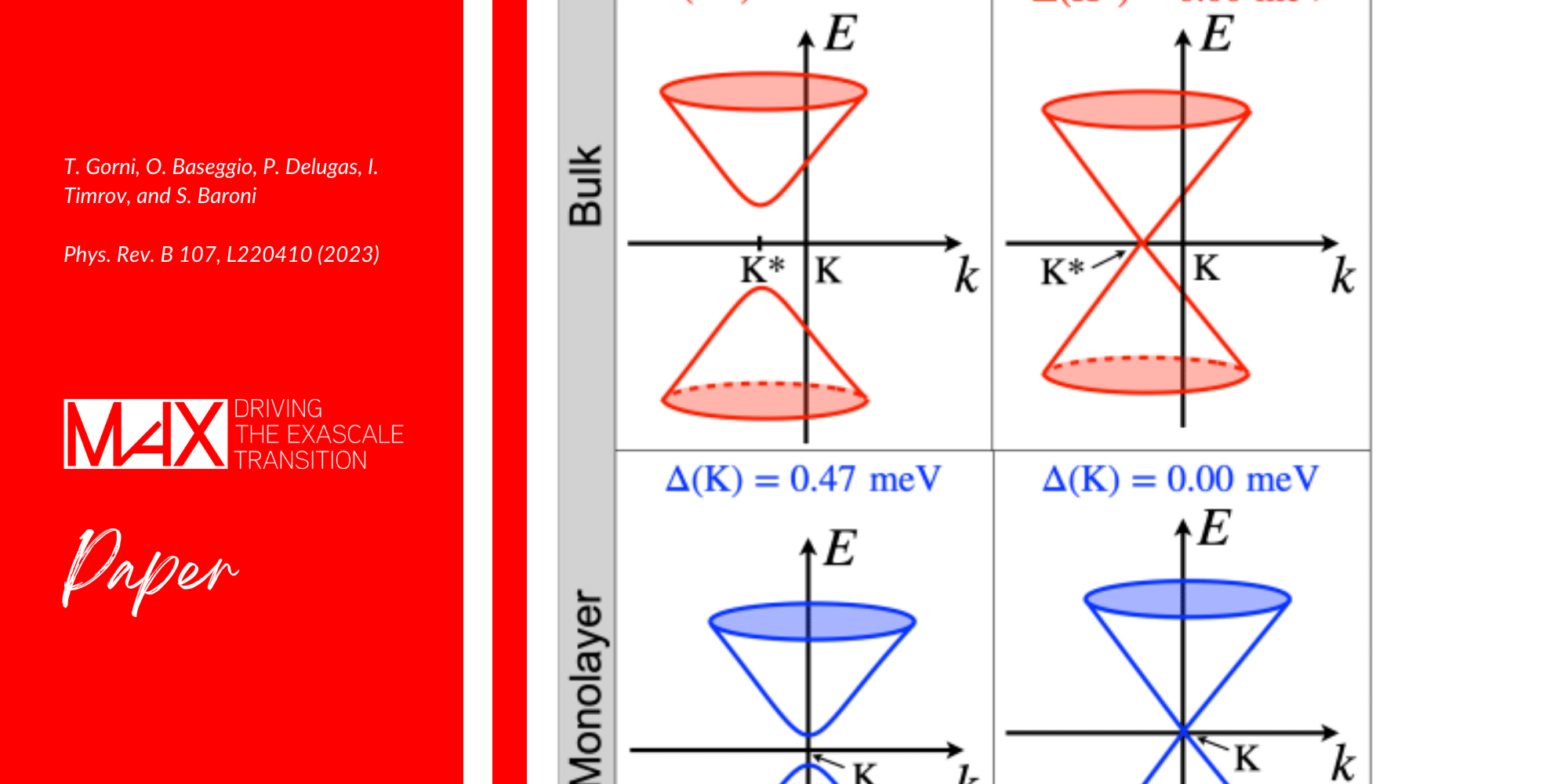
At the zone border in the spin excitation spectrum of CrI3 quasi-two-dimensional single crystals a gap is observed whose nature is still controversial. In 2017 convincing evidence that a single layer of CrI3 was magnetically ordered came from MOKE measurements, making monolayer CrI3 the first two-dimensional intrinsic ferromagnet ever reported.
By performing first-principles calculations based on time-dependent density functional perturbation theory, a team of researchers show that the observed gap results from a combination of spin-orbit and interlayer interaction effects.
<< Precise measurements of the magnetic properties of such a system, consisting of a single layer of Cr and I atoms, is a formidable challenge>> says Tommaso Gorni (CINECA), first author of the paper. << Inelastic neutron scattering spectroscopy could not fully clarify the size and origin of an unexpected gap found in its magnetic excitation spectra, which ultimately determines how spin waves travel within the material. By means of atomistic first-principles simulations, we simulated the magnetic excitation spectrum of CrI3 crystals in different configurations, specifically for a single CrI3 layer and for several multilayer configurations with different interlayer distances>> continues Gorni.
By comparing the results of their simulations with the experiments, the team of researchers could provide evidence that the gap in the magnetic excitation spectrum of CrI3 is almost entirely due to the interaction between different atomic layers. This predicts the absence of such a gap in the excitation spectrum of a single CrI3 layer, a measurement currently beyond the experimental resolution.
To carry out the simulations with the proper degree of accuracy, the team relied on state-of-the-art algorithms, as the one implemented in the turboMagnon code of the Quantum ESPRESSO distribution, to take into account the effects of special-relativity laws in the quantum-mechanical modelling of the compound.
About Quantum ESPRESSO
Quantum ESPRESSO is an integrated suite of open-source computer codes for electronic-structure calculations and materials modeling at the nanoscale. It is based on density-functional theory, plane waves, and pseudopotentials. The codes of the suite may perform many types of ground-state calculations: self-consistent energies, forces and stresses, structural optimization, molecular dynamics (PW and CP); search for transition pathways (NEB). The suite also contains a rich apparatus of routines and methods for the computation of the linear response to external perturbations.
Reference article:
First-principles study of the gap in the spin excitation spectrum of the CrI3 honeycomb ferromagnet.