
27 May 2024
Investigating adsorption behaviours induced by chemical functionality replacement towards electro-galvanised steel corrosion.

Using both experimental and theoretical approaches, a team of researchers presented new insights for rational inhibitor engineering, enabling the realization of tailored corrosion inhibition properties.
Global efforts and initiatives to curb greenhouse gas emissions have been propelled by the increased environmental awareness of these last years. In this framework, implementing efficient and sustainable methods for metal corrosion protection mitigates the carbon footprint of the metal-producing industry.
<<Using corrosion inhibitors has emerged as a well-documented strategy for effectively mitigating metal corrosion in aggressive environments. Hence, a deep understanding of corrosion inhibitor behaviors at the metal-aqueous interface linked to unique molecular features is crucial for effective metal protection>> says José María Castillo Robles (RMIT University), one of the authors of the paper.
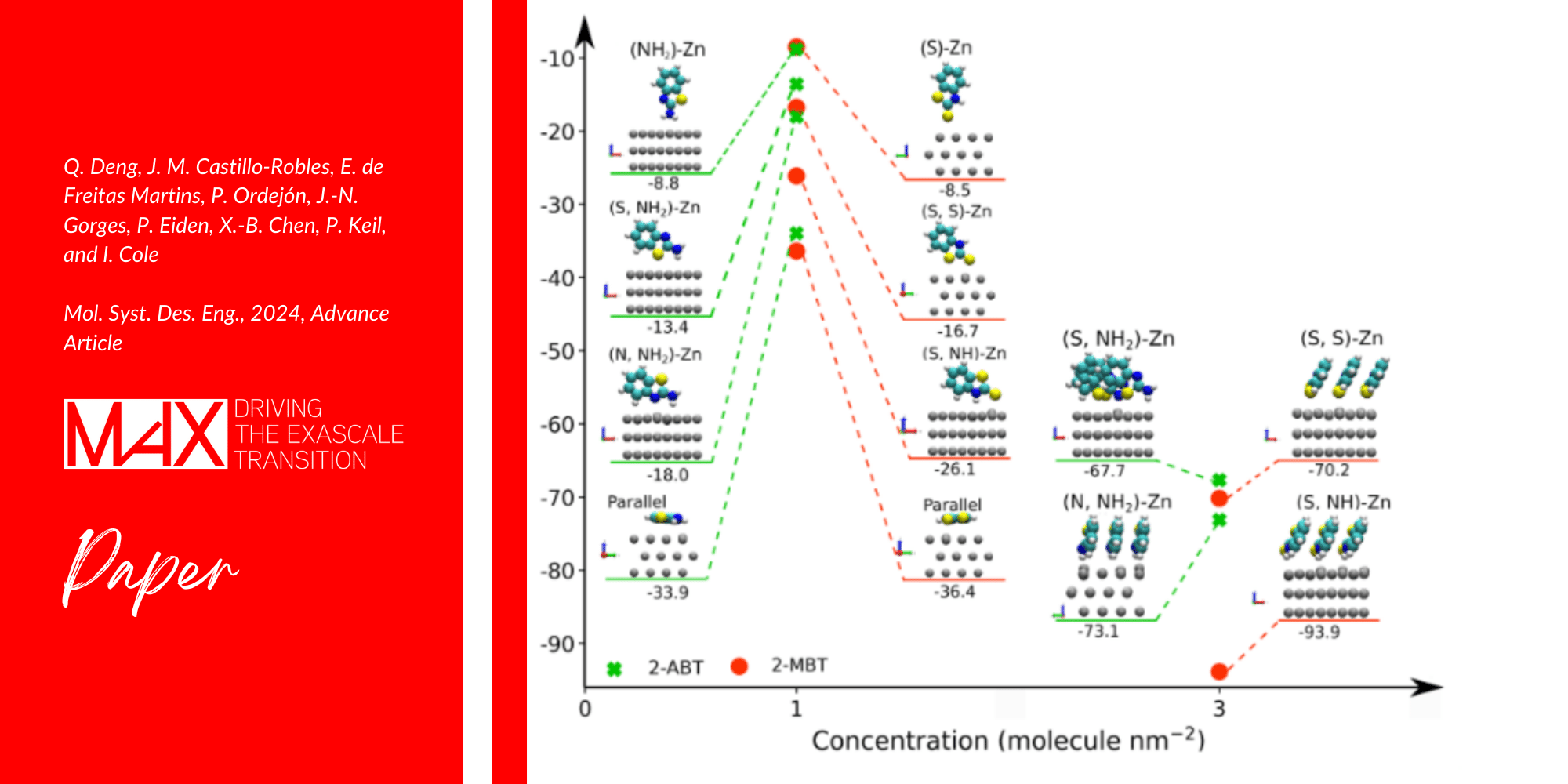
This study sheds light on tailored inhibition for electro-galvanized steel corrosion, exploring distinct adsorption patterns of benzothiazole derivatives (2-mercaptobenzothiazole and 2-aminobenzothiazole) using both experimental and theoretical approaches.
To perform Density Functional Theory (DFT) simulations, the team of researchers used the SIESTA package: <<Thanks to the functionalities of SIESTA, our study could reveal that high concentrations of inhibitor molecules tend to form a packed self-assembled monolayer (SAM) on the surface, where the presence heteroatoms enhance surface–molecule interaction>> continues Castillo Robles.
<<Additionally, DFT suggests that the strong binding strength of 2-MBT could facilitate the formation of complexes with displaced Zn. SIESTA, which uses a basis set of strictly localized atomic orbitals and norm-conserving pseudopotentials, allows us to model systems of dozens to hundreds of atoms with a modest cost and high accuracy, being crucial for achieving these results.>> concludes Castillo Robles.
About SIESTA
SIESTA is a first-principles materials simulation program based on density-functional theory (DFT). SIESTA is known to be used in a wide range of applications, encompassing materials science, nanotechnology, catalysis, biological sciences (including interaction between organic and inorganic materials), geology and materials under high pressure, Martian geochemistry, materials for nuclear reactors, and astrophysical and atmospheric systems.
Reference article:
Inhibitory behaviour and adsorption stability of benzothiazole derivatives as corrosion inhibitors towards galvanised steel.